
FUENTE: Tamayo JM. "Psicofarmacologia On-Line" http://psicofarmacologia.info/. Jorge M Tamayo, psiquiatra y farmacólogo clínico de la Universidad de Antioquia (Medellín, Colombia).
Ansiolíticos, sedantes e hipnóticos
BENZODIACEPINAS
Las BZD están entre las medicinas más corrientemente prescritas en el mundo. En el año 1985, se prescribieron 61 millones de recetas en los Estados Unidos (4% de todas las prescripciones), encontrándose seis BZD entre los 25 productos más prescritos (Ruiz, 1992).
FARMACOCINÉTICA
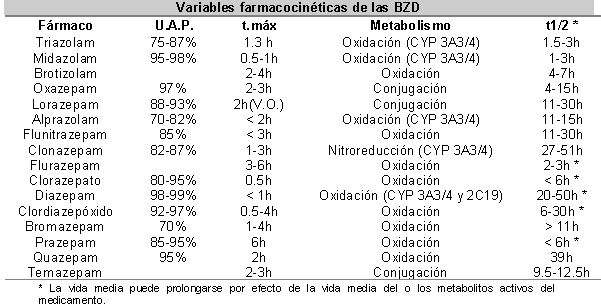{kind=link}
· Tienen metabolismo hepático (solicitar pruebas hepáticas previas). Aunque pueden provocar inducción enzimática, este efecto es de poca significancia clínica en humanos.
· Se absorben bien por V.O. alcanzando picos plasmáticos entre los 30 minutos y las 8 horas.
· Alta biodisponibilidad y liposolubilidad.
· Atraviesan fácilmente la barrera hematoencefálica por su alta liposolubilidad (DeVane et al., 1991).
· Usar por vía I.V. en casos muy específicos.
· En caso de vía I.M., utilizar el músculo deltoides; las únicas BZD que tienen una absorción predictible por esta vía son el Midazolam y el Lorazepam.
· En sujetos obesos o en ancianos su actividad puede prolongarse por la unión a tejido graso y disminución del metabolismo hepático respectivamente (Harvey, 1985).
· Los niveles plasmáticos tienen poca correlación con la respuesta terapéutica. Esto sólo ha podido demostrarse con Alprazolam, aunque un estudio encontró pérdida de tal correlación al cabo de 8 semanas de administración (Lesser et al., 1992; Greenblatt et al., 1993).
FARMACODINAMIA
Facilitan la transmisión gabaérgica y disminuyen el recambio de algunos neurotransmisores como noradrenalina, serotonina, acetilcolina y dopamina, lo que contribuye a su efecto sedativo y ansiolítico (Harvey, 1985). Al incrementar la actividad del receptor de BZD (sitio-w), estrechamente en contacto con el complejo iónico GABAA, permiten una mayor activación de los canales de Cl- por el GABA o sus agonistas (muscimol, p.ej.), permitiendo que el ion fluya al interior de la membrana, inhibiendo la excitabilidad neuronal. También se ha sugerido un incremento en la concentración de Ca++ intraneuronal dependiente de la conductancia de K+ (Tallman et al., 1980).
La modulación del complejo iónico GABAA por parte de las BZD provocan cambios en la actividad eléctrica cerebral: en la vigilia, disminuyen las ondas alfa e incrementan las delta (efecto hipnótico) y las beta (principalmente en áreas frontal y rolándica (diferente a los barbitúricos). En el sueño, incrementan la actividad beta, disminuyen la latencia de inicio y el número de despertares, disminuyen las fases 1, 3, 4 y la cantidad total de sueño REM e incrementan la fase 2 y el número de ciclos de sueño REM. Al ser suspendidas abruptamente pueden llevar a un efecto de rebote con incremento de la fase REM y presentación de pesadillas y sueños extremadamente bizarros (Ballenger, 1995).
La modulación del complejo iónico GABAA por parte de las BZD provocan cambios en la actividad eléctrica cerebral: en la vigilia, disminuyen las ondas alfa e incrementan las delta (efecto hipnótico) y las beta (principalmente en áreas frontal y rolándica (diferente a los barbitúricos). En el sueño, incrementan la actividad beta, disminuyen la latencia de inicio y el número de despertares, disminuyen las fases 1, 3, 4 y la cantidad total de sueño REM e incrementan la fase 2 y el número de ciclos de sueño REM. Al ser suspendidas abruptamente pueden llevar a un efecto de rebote con incremento de la fase REM y presentación de pesadillas y sueños extremadamente bizarros (Ballenger, 1995).
Los receptores de BZD son de dos tipos : los w1, con alta afinidad por las betacarbolinas (péptidos endógenos) y las triazolopiridazinas como Quazepam, Halazepam y Zolpidem. Son los más comunes en el cerebelo y corteza cerebral y participan en la mediación del sueño. Los w2 influyen en la cognición, memoria y control motor. Se hallan principalmente a nivel de la corteza (acción anticonvulsivante), hipocampo y amígdala (acción ansiolítica), y en menor cantidad en el tálamo y base del cerebro (acción sedativa).
Además actúan sobre los receptores presentes en el eje hipotálamo-hipófiso-adrenal, inhibiendo la secreción de ACTH, cortisol, TSH y prolactina, que se liberan en respuesta al estrés. En el cerebelo conducen a ataxia y relajación muscular (también por efecto medular); y en el procencéfalo e hipocampo tienen efectos sobre la memoria (Zorumski & Isenberg, 1991). Últimamente se ha estado trabajando en el desarrollo de agonistas parciales que posean efectos ansiolíticos, pero sin sedación ni síndrome de abstinencia como es el caso del Abecarnil (Ballenger, 1991).
Además actúan sobre los receptores presentes en el eje hipotálamo-hipófiso-adrenal, inhibiendo la secreción de ACTH, cortisol, TSH y prolactina, que se liberan en respuesta al estrés. En el cerebelo conducen a ataxia y relajación muscular (también por efecto medular); y en el procencéfalo e hipocampo tienen efectos sobre la memoria (Zorumski & Isenberg, 1991). Últimamente se ha estado trabajando en el desarrollo de agonistas parciales que posean efectos ansiolíticos, pero sin sedación ni síndrome de abstinencia como es el caso del Abecarnil (Ballenger, 1991).
INDICACIONES GENERALES
· Endoscopias, cateterismos
· Ansiedad generalizada (BZD de vida media intermedia o larga y de inicio de acción rápido).
· Preanestesia y postquirúrgicos
· Síndromes convulsivos (epilepsia) y status epiléptico (Diazepam)
· Relajantes musculares (Diazepam)
· Alcoholismo agudo (fase de abstinencia)
· Inducción y/o mantenimiento del sueño (en insomnio por ansiedad, con BZD de vida media corta y de rápido inicio de acción; también útiles en pesadillas, terrores nocturnos y sonambulismo por la disminución de la fase 4).
· Ansiedad anticipatoria en Trastorno de pánico (Alprazolam, Clonazepam, Lorazepam, Diazepam).
· Ansiedad por retirada del respirador
· Reacción aguda al estrés
· síndromes dolorosos de corta duración
· Enfermedades psicosomáticas (colon irritable)
· Agitación senil psicomotriz
· Delirium (como auxiliar de los antipsicóticos : Diazepam, Clordiazepóxido o Lorazepam I.V.)
· Reacciones distónicas agudas o mioclónicas (Diazepam, Clonazepam).
· Infarto agudo del miocardio, Diazepam, 2-10 mg, 2 a 4 veces al día (disminuye la excitación simpática protegiendo al paciente de la arritmia cardíaca súbita).
· Agitación de cualquier naturaleza (manía, psicosis...), Lorazepam I.M., Clonazepam.
EVENTOS ADVERSOS
· Laxitud muscular
· A dosis convencionales no producen efectos importantes en el sistema cardiorespiratorio; incluso en sobredosis suelen ser seguras y no producen marcada depresión respiratoria (por el incremento compensatorio del volumen corriente) siempre y cuando no se combinen con otros depresores del SNC como el alcohol (Greenblatt et al., 1977 ; Finkle et al., 1979). La depresión respiratoria y/o hipotensión y taquicardia se presentan más que todo con dosis preanestésicas de Midazolam, especialmente si se combina con grandes dosis de Fentanilo.
· Disminución de la agudeza mental y de las habilidades motoras (Las BZD se asocian a un riesgo 4.9 veces mayor de accidentalidad automotriz que el de los sujetos controles) (Skegg et al., 1979).
· Agitación paradójica (Diazepam, Clordiazepóxido)
· Sedación, somnolencia
· Potenciación de los efectos de barbitúricos o alcohol o antidepresivos.
· Púrpura no trombocitopénica
· Fotosensibilidad (visión borrosa)
· Despertar prematuro con insomnio de rebote (Triazolam, Midazolam).
· Ataxia con dosis altas o en ancianos y mayor riesgo de caídas (el riesgo se incrementa en 1.8 veces). Las BZD de vida media corta no exhiben este riesgo (Ray et al., 1987), pero se asocian a un significativo deterioro de las habilidades psicomotoras (Smiley, 1987).
· Pueden ser teratogénicas (paladar hendido)
· Alergias cutáneas y cefaleas
· La amnesia anterógrada (relacionada con aspectos biográficos) se presenta 1 a 3 horas después de la toma de BZD de acción corta principalmente como en el caso del Triazolam por compromiso de la transferencia de datos a las áreas corticales hipocámpicas donde son almacenados (memoria episódica), sin compromiso de la memoria inmediata, ni de la retrógrada; (Rosser, 1988; Greenblatt et al., 1991).
· Presentan menor riesgo de abuso y dependencia que los barbitúricos. Las BZD de vida media larga prácticamente no presentan fenómeno de tolerancia y el riesgo de abuso es mínimo, pero pueden acumularse (Rosser, 1988).
Estos fenómenos no parecen deberse a un cambio en el número de receptores para BZD en el complejo iónico GABAA (down-regulation), sino más bien a una desensibilización de los mismos (Gallager et al., 1984). El riesgo de abuso y dependencia es mayor en BZD de vida media corta administradas a dosis elevadas. Lorazepam, Alprazolam, Triazolam y Diazepam son de mayor riesgo que Oxazepam, Clorazepato y Clonazepam (Griffiths et al., 1990). Aunque un estudio en pacientes que consumían Lorazepam y Alprazolam por más de tres meses para trastornos de ansiedad diversos no reveló síndrome alguno de abstinencia en el proceso de descontinuación (Romach et al., 1995), un estudio a largo plazo (1 año) en usuarios de BZD con suspensión abrupta al cabo de dicho lapso, reveló que un 57% de aquellos que consumían BZD de vida media corta presentaron síntomas de abstinencia contra un 27% de los que consumían BZD de vida media larga (Rickels et al., 1990). Los síntomas de abstinencia se caracterizan por ansiedad, insomnio, fotosensibilidad, audiosensibilidad, taquicardia, hipertensión sistólica leve, temblor, cefalea, sudoración, dolor abdominal o convulsiones (Roy-Byrne & Hommer, 1988).
· Presentan menor riesgo de abuso y dependencia que los barbitúricos. Las BZD de vida media larga prácticamente no presentan fenómeno de tolerancia y el riesgo de abuso es mínimo, pero pueden acumularse (Rosser, 1988).
Estos fenómenos no parecen deberse a un cambio en el número de receptores para BZD en el complejo iónico GABAA (down-regulation), sino más bien a una desensibilización de los mismos (Gallager et al., 1984). El riesgo de abuso y dependencia es mayor en BZD de vida media corta administradas a dosis elevadas. Lorazepam, Alprazolam, Triazolam y Diazepam son de mayor riesgo que Oxazepam, Clorazepato y Clonazepam (Griffiths et al., 1990). Aunque un estudio en pacientes que consumían Lorazepam y Alprazolam por más de tres meses para trastornos de ansiedad diversos no reveló síndrome alguno de abstinencia en el proceso de descontinuación (Romach et al., 1995), un estudio a largo plazo (1 año) en usuarios de BZD con suspensión abrupta al cabo de dicho lapso, reveló que un 57% de aquellos que consumían BZD de vida media corta presentaron síntomas de abstinencia contra un 27% de los que consumían BZD de vida media larga (Rickels et al., 1990). Los síntomas de abstinencia se caracterizan por ansiedad, insomnio, fotosensibilidad, audiosensibilidad, taquicardia, hipertensión sistólica leve, temblor, cefalea, sudoración, dolor abdominal o convulsiones (Roy-Byrne & Hommer, 1988).
INTERACCIONES MEDICAMENTOSAS
· Síndrome de apnea obstructiva del sueño
· Miastenia gravis
· Insuficiencia respiratoria
· Hepatopatía (disminuir la dosis).
· Insuficiencia renal (disminuir la dosis en un tercio por disminución de la albúmina).
· Glaucoma de ángulo estrecho
· Embarazo y lactancia (Categoría D)
· Menores de 18 años (?)
· Está demostrado que los alcohólicos y sus parientes cercanos tienen un mayor riesgo de abuso de BZD (tolerancia cruzada) y por ello debe evitarse la prescripción de estos psicofármacos en tales pacientes (Ciraulo et al., 1989).
· Uso concomitante con Cimetidina o Disulfiram pues disminuyen el metabolismo de las BZD (Ruffalo & Thompson, 1980); anticonceptivos orales, Isoniazida, Fluoxetina, sustancias psicoestimulantes, Propranolol, Cloranfenicol, Alopurinol, que incrementan la vida media (disminuir las dosis de la BZD) (Rosser, 1988).
MEDICAMENTOS
Para iniciar tratamiento, dar la mitad de la dosis promedio e ir incrementando en un 25% la dosis cada 2 días hasta la mejoría. Revisar cada 2 a 4 meses.
Para suspender, disminuir el 25% cada 3 días. La interrupción brusca puede dar ataxia, parestesias, depresión, insomnio, despersonalización e incremento de la sensibilidad a estímulos. Las BZD de vida media corta se deben dar en forma intermitente al suspender la administración continua (Shader & Greenblatt, 1993).
Por todo lo anterior, deben darse las dosis más bajas posibles, evitando utilizarlas como hipnóticos por más de 10 noches y como ansiolíticos por más de 5 semanas, en especial si se utiliza Alprazolam, Triazolam o Lorazepam y en pacientes con Trastorno de pánico (uso crónico) o con trastorno de personalidad pasivo-agresiva, histriónica, somatoforme o asténica.
Las BZD son clasificadas en tres grupos según la duración de su acción. Se mencionarán sólo aspectos no contemplados en los apartes anteriores :
1. De acción corta:
Son las BZD que más posibilidad tiene de desarrollar síndrome de abstinencia. Su vida media en promedio es inferior a 5 horas; suelen implicarse en intentos suicidas y poseen metabolitos activos.
Triazolam |
farmacocinética:
Metabolito activo por acción de la CYP3A3/4 : alfa hidroxitriazolam (vida media = 3-8 h).
farmacodinamia:
Rápido inicio de acción; no altera el sueño REM; disminuye la fase 4, pero incrementa el tiempo total de sueño disminuyendo el número de despertares durante el mismo.
indicaciones:
Hipnótico. Preanestésico.
dosis:
0.125-0.5 mg/día
precaución:
Mayores reacciones paradojales (insomnio de rebote) y amnesia anterógrada. No debe darse concomitantemente con Ketoconazol o Itraconazol debido a la competición enzimática (CYP3A) que podría conducir a un incremento de los niveles plasmáticos de los azoles y por ende a un alto riesgo de arritmias cardíacas (Varhe et al., 1994).
Midazolam |
farmacocinética:
Alta liposolubilidad. Metabolito activo por hidroxilación (CYP3A3/4): alfa-hidroximidazolam (vida media = 1 h).
indicaciones:
Hipnótico potente, preanestésico.
dosis:
3.25-15 mg/día; 0.15-0.2 mg/kg (niños)
precaución:
Puede dar amnesia anterógrada. No debe darse concomitantemente con Ketoconazol, Itraconazol o Rifampicina debido a la competición enzimática (CYP3A3/4) que podría conducir a un incremento de los niveles plasmáticos de los azoles y por ende a un alto riesgo de arritmias cardíacas o a la disminución significativa de los niveles plasmáticos y del efecto terapéutico de la BZD por la administración de Rifampicina concomitante (Olkkola et al., 1994 ; Backman et al., 1996).
Brotizolam |
farmacocinética:
Metabolito activo: 1-metilhidroxibrotizolam.
farmacodinamia:
No modifica las etapas 3 y 4 del sueño NREM. Aparentemente sin modificación del sueño REM (Kim, et al., 1993).
indicaciones:
Hipnosedante; preanestesia.
dosis:
1/2 a 1 comprimido al acostarse.
2. De acción intermedia:
Tienen una vida media promedio de 6 a 12 horas.
Oxazepam |
farmacocinética:
Inicio de acción lento
indicaciones:
Ansiolítico; de elección en ancianos y pacientes con trastornos renales o hepáticos.
dosis:
30-60 mg/día en 3 tomas.
Lorazepam |
farmacocinética:
Metabolito inactivo: glucurónido.
indicaciones:
Ansiolítico potente; puede ser útil en Trastorno de pánico; seguro en ancianos, nefrópatas y hepatópatas, pero presenta grandes riesgos de síndrome de abstinencia.
dosis:
0.5-6 mg/día (2 mg) V.O. o S.L. (0.05 mg/kg/día). Máximo 10 mg I.M. (absorción rápida constante = 1.5 horas) o I.V.
Alprazolam |
farmacocinética:
Metabolito: alfa-hidroxialprazolam por la 4-hidroxilación llevada a cabo a través de la CYP3A3/4. Vida media = 10-12 horas.
indicaciones:
Trastorno de pánico (solo o asociado) (Chouinard et al., 1982 ; Ballenger et al., 1988 Schweizer et al., 1988 ; Charney & Woods, 1989); ansiolítico potente (Munjack et al., 1989); ansiedad asociada con síntomas depresivos (sensibiliza los receptores 5-HT2) (Hoehn-Saric et al., 1988).
dosis:
En ansiedad, 0.25-0.5 mg 3 veces / día; en trastorno de pánico, 1.5-6 mg/día (máximo 8 mg) divididos en 3 a 4 tomas.
precauciones:
Debido a su metabolismo por la CYP3A3/4, debe tomarse con precaución en paciebntes que reciben concomitantemente Ketoconazol, Terfenadina, macrólidos o Nefazodone.
Temazepam |
indicaciones:
Hipnótico con sedación residual mínima (leve en las mañanas). Imita el sueño fisiológico y por ello se acerca al hipnótico ideal.
dosis:
15-30 mg/día.
Flunitrazepam |
farmacocinética:
Metabolito activo: N-desmetilnitrazepam (vida media = 31 horas; efecto hipnótico = 8 horas). Metabolito inactivo: 7-aminoflunitrazepam (vida media = 23 horas).
indicaciones:
Hipnótico, sedante, preanestésico.
dosis:
1-4 mg/día (máximo 6) por máximo 4 semanas. En ancianos utilizar 0.5 mg.
precauciones:
Amnesia anterógrada, pero sin síntomas de abstinencia.
Clonazepam |
farmacocinética:
Tiene una biodisponibilidad oral del 80%. Puede generar autoinducción moderada. Semetaboliza por nitrorreducción, lo que permite su administración con la mayoría de psicofármacos sin que se presentes efectos de acumulación (Greenblatt et al., 1992). Utiliza el mismo metabolismo del Nitrazepam, el cual ha mostrado ser seguro en pacientes ancianos o con cirrosis hepática (Greenblatt et al., 1985).
farmacodinamia:
Efecto GABA modulador al unirse a las subunidades específicas del receptor GABAA.
indicaciones:
Hipnótico, sedante potente, anticomicial; Trastorno de pánico (solo o asociado) a dosis de 3-6 mg/día; Trastorno bipolar resistente (profilaxis y episodio maníaco) a dosis de 2-16 mg/día; Trastorno de Tourette, detoxificación de BZD, T.O.C., mioclonías nocturnas, agitación, agresividad (Chouinard et al., 1983) y discinesia tardía.
dosis:
1.5-10 mg/día (máximo 20) en 2 a 3 tomas (iniciar con 0.5 mg c/12 horas) (0.07-1 mg./kg./día). Niveles plasmáticos en epilepsia mioclónica = 15-75 ngr./ml.
efectos adversos:
Ataxia, sedación, disfunción sexual.
precauciones:
Evitar su uso concomitante con IMAO's, anticoagulantes y anticonvulsivantes.
3. De acción prolongada:
Vida media promedio de más de 12 horas (50-120 horas por sus metabolitos). Tienden a acumularse con repetidas dosis, lo que incrementa el riesgo de sedación diurna y disminución de concentración y memoria. Suelen ser de baja potencia.
Flurazepam |
farmacocinética:
Profármaco. Metabolitos activos: N-hidroxietilflurazepam (vida media = 2 horas) y N-desalquilflurazepam (vida media = 47-100 horas).
farmacodinamia:
El porcentaje de sueño REM se disminuye, con un incremento en la etapa 2 del NREM y reducción de la 3 y 4.
indicaciones:
Hipnótico.
dosis:
15-45 mg/día (30)
precauciones:
Como hipnótico, el efecto puede prolongarse en el día. Usar con cuidado en ancianos. Síndrome de retiro menos severo.
Clorazepato |
farmacocinética:
Profármaco. Metabolito activo: Desmetildiazepam (vida media = 30-210 horas).
indicaciones:
Ansiolítico
dosis:
20 mg/día (10-50).
precauciones:
No usar en pacientes con aclorhidria, ni anemia perniciosa, ni con antiácidos.
Diazepam |
farmacocinética:
Altamente liposoluble (alcanzando niveles sanguíneos elevados en forma rápida y concentraciones estables en 5-10 días - efecto de acumulación) al igual que sus metabolitos activos: desmetildiazepam (por N-demetilación) y Temazepam (vida media = 10 horas) (por 3-hidroxilación). El desmetildiazepam, a su vez, se convierte por 3-hidroxilación en Oxazepam (vida media = 4-15 horas). El aclaramiento plasmático del Diazepam y el desmetildiazepam es más bajo en metabolizadores pobres de S-mefenitoína que en metabolizadores rápidos. Así, la N-demetilación del Diazepam y la hidroxilación del desmetildiazepam son catalizadas por la CYP2C19 en un 57%, mientras la CYP3A3/4 se encarga del 43% restante (Schmider et al., 1996). La hidroxilación del Diazepam a Temazepam es catalizada por la CYP3A3/4 (Bertilsson et al., 1989). La CYP1A2 también está implicada en el metabolismo del Diazepam, toda vez que su administración concomitante con Fluvoxamina disminuye su aclaramiento (Perucca et al., 1994).
indicaciones:
Relajante muscular; en tratamiento de supresión de drogas psicotrópicas. Anticomicial. Ansiolítico. Trastorno de pánico (Noyes et al., 1984).
dosis:
5-40 mg/día como ansiolítico en 2 a 4 tomas. 10 mg / día como hipnótico. La administración I.M. o I.V. puede repetirse cada 3 a 4 horas; la dosis en niños es de 0.04-0.2 mg / kg.
Clordiazepóxido |
farmacocinética:
Primera BZD. Metabolitos activos: Desmetildiazepóxido (vida media = 10-18 horas); Demoxepam (vida media = 14-95 horas) y Desmetildiazepam. Poco potencial de abuso por inicio lento de acción.
indicaciones:
Ansiolítico. Se ha descrito alguna utilidad en depresiones ansiosas, asociado a Amitriptilina, 10 mg/día. Útil en detoxificación alcohólica.
dosis:
15-100 mg/día. En caso de síntomas de abstinencia severos, 50-100 mg I.V. cada 2 a 4 horas si es necesario.
Bromazepam |
farmacocinética:
Metabolito activo: 3-hidroxibromazepam (vida media mayor de 20 horas).
indicaciones:
Ansiolítico (sin ser hipnótico, ni miorelajante); fobias y agresión (0.15 mg/kg).
dosis:
1.5-12 mg/día
Prazepam |
farmacocinética:
Profármaco: se convierte en Desmetildiazepam (pico máximo de concentración sérica a las 6 horas de administración)
indicaciones:
Ansiolítico
dosis:
10-60 mg/día
Quazepam |
farmacocinética:
Metabolitos activos: 2-oxoquazepam y N-dealquil-2-oxoquazepam (desalquilflurazepam) (vidas medias = 39 y 73 horas respectivamente).
indicaciones:
Hipnótico
dosis:
7.5-15 mg / día
GABAÉRGICOS NO BENZODIACEPÍNICOS
Zopiclona |
farmacocinética:
Absorción rápida (90%).La vida media puede incrementarse en 2 a 3 horas más en los ancianos y puede llegar a ser de 11 horas en cirróticos. Metabolitos hepáticos inactivos. Excreción renal (75%).
farmacodinamia:
Hipnótico no BZD del tipo de las ciclopirrolonas que se fija a los receptores BZD w-1 y w-2 del cerebro, con poca somnolencia diurna, similar al placebo e inferior a la producida por el Temazepam. Produce disminución de la latencia del sueño y de la etapa 1 menor que la producida por BZD (Fleming et al., 1988). Incrementa el tiempo total de sueño, sin afectar la arquitectura de las etapas 3 y 4. Aumenta la latencia del REM y disminuye su duración (Kim et al., 1993).
efectos adversos:
Puede dar trastornos de memoria anterógrada durante el primer día de tratamiento. Potencia la acción sedante-hipnótica de los barbitúricos, alcohol y BZD. No produce síntomas de abstinencia aún después de administraciones superiores a 3 semanas. No afecta la función respiratoria central o periférica. Los efectos adversos más frecuentemente reportados fueron : Náusea - vómito (4.2%), Boca seca (8.4%), Sabor amargo (15.1%).
dosis:
3.75-7.5 mg/día (4.5 mg) en la noche. Sin diferencias en su efecto hipnótico cuando se compara con Nitrazepam, Flunitrazepam, Triazolam o Temazepam (Wheatley, 1985).
Zolpidem |
farmacocinética:
Tiene un rápido comienzo de acción (superior al del Triazolam y Temazepam) (Rush & Griffiths, 1996).
farmacodinamia:
Es una imidazopiridina no relacionada con las BZD, pero se une selectivamente a los sitios w-1 de los receptores benzodiacepínicos. No produce tolerancia ni insomnio de rebote e incrementa las fases II, III y IV sin afectar el sueño REM (Shlarf et al., 1994).Sin embargo, un estudio comparativo con Triazolam y Temazepam reveló similares efectos sobre el aprendizaje, memoria y ejecución a estos compuestos (Rush & Griffiths, 1996).
indicaciones:
Insomnio transitorio (máximo por 20 días) y geriátrico. Su uso prolongado puede estar asociado a los mismos inconvenientes de las benzodiacepinas. Un estudio comparativo con Alprazolam en 122 pacientes con trastorno de ansiedad generalizada, mostró una menor presentación de síndromes de abstinencia a dosis de 150 mg./día (divididos en 3 tomas) (p = 0.044). El medicamento en general fue bien tolerado (Frattola et al., 1994).
dosis:
10 a 20 mg en la noche.
precauciones:
Puede llevar a sedación residual al día siguiente y potenciar los efectos sedantes del alcohol.
Amobarbital sódico |
farmacocinética:
Como todos los barbitúricos, se deriva del ácido barbitúrico (2,4,6-trioxohexahidropirimidina). Se absorbe rápidamente por estómago cuando se administra por vía oral. Se metaboliza a nivel hepático por oxidación, produciendo metabolismos inactivos que se excretan rápidamente por orina. Puede llevar a la inducción enzimática cuando se administra en forma crónica (Rall, 1993).
farmacodinamia:
Incrementa los procesos inhibitorios del SNC mediados por el GABA, con acción sobre el sistema reticular activante mesencefálico. No desplaza a las BZD de su receptor, incrementa su afinidad por el mismo, ya que actúa directamente sobre el ionóforo de Cl- dependiente de receptores GABAA incrementando la duración de su apertura (a diferencia de las BZD que incrementan sólo la frecuencia de apertura) (Nishino et al., 1995). Disminuye la latencia del sueño, incrementa la etapa 2 y disminuye las etapas 3 y 4. Prolonga además, la latencia del REM y disminuye el número de ciclos y la duración total del mismo.
indicaciones:
Hipnótico (actualmente de poco uso para esta indicación) y preanestésico; induce el sueño en un tiempo de 20-60 minutos, con un efecto que dura unas 4 horas. Puede ser usado parenteralmente para producir una relajación que facilite la entrevista psiquiátrica, para permitir la disminución del trastorno por experiencias negativas previas y es útil en el diagnóstico de catatonía al conducir a la movilización de estos pacientes (McCall et al., 1992).
dosis:
65-200 mg antes de dormir; 6 mg/kg/día en niños. En la forma parenteral (I.M. o I.V.) pueden utilizarse hasta 500 mg.
efectos adversos:
Por ser un supresor de la respiración a nivel del tallo cerebral, puede causar depresión respiratoria fatal a dosis terapéuticas en pacientes con apnea del sueño. Se contraindica en pacientes con porfiria, por incrementar la síntesis de porfirina como todos los barbitúricos. Se requiere gran precaución en pacientes con trastornos hepáticos por la posibilidad de llevar a una sobredosis. En pacientes consumidores crónicos, la suspensión abrupta puede llevarlos a presentar ansiedad, agitación, temblor y convulsiones. En caso de sobredosis (10 veces la dosis hipnótica usual) la muerte puede ocurrir en un 0.5% a 10% de los casos (Rall, 1993). Puede producir disforia paradójica, hiperactividad y alteración cognitiva. Como todos los barbitúricos, difiere de las BZD en su alto poder adictivo, alto grado de tolerancia y dependencia y su bajo índice terapéutico. Puede afectar el metabolismo de los anticoagulantes, anticonvulsivantes, corticoides, ACTH, estrógenos y progesterona (anticonceptivos orales), antidepresivos heterocíclicos.
Meprobamato |
farmacocinética:
Perteneciente a los carbamatos. Baja unión a proteínas plasmáticas. La vida media inicial se incrementa a 24 a 48 horas con la administración crónica.
farmacodinamia:
Causa depresión del SNC más no anestesia, llevando a suprimir comportamientos en respuesta a eventos adversos previos en animales. Deprime los reflejos espinales polisinápticos más que los monosinápticos (acción más selectiva que a de los barbitúricos) (Rall, 1993). Tiene propiedades anticonvulsivantes similares a la Etosuximida, pudiendo agravar las convulsiones tónico-clónicas generalizadas. No se conocen receptores específicos para su acción; se postula que puede actuar a través de la inhibición del complejo BZD-GABA-ionóforo de cloro, pero al parecer su unión al receptor BZD es cinco veces menor de lo requerido para la inhibición competitiva con las BZD. Otros postulan que la acción central del Meprobamato está asociada con la potenciación de la liberación de adenosina endógena, al inhibir su recaptación; esto explicaría los efectos depresores del SNC y de los reflejos espinales. Finalmente, también se ha postulado su acción sobre el recambio de noradrenalina a nivel cerebral, disminuyen el estrés asociado al incremento de ésta (Cole & Yonkers, 1995).
indicaciones:
Ansiolítico, comparable a las BZD y superior al placebo (Rickels et al., 1988).
dosis:
400 mg a la hora de acostarse (hipnótico); iniciar con 200 - 400 mg / día e incrementar en caso de ser necesario. La dependencia física se ha observado con dosis tan bajas como 3200 mg / día. Actualmente se deja como último recurso cuando las BZD u otros ansiolíticos han fallado (Cole & Yonkers, 1995).
precauciones:
Al formar un bezoar a nivel del estómago, en caso de sobredosis el paciente puede presentar fluctuaciones del estado de conciencia por disolución del bezoar. La dosis letal es de 12.000 mg (30 tabletas) (Rall, 1993). Mayor riesgo de adicción que las BZD y los barbitúricos. El síndrome de abstinencia se caracteriza por ansiedad, intranquilidad, debilidad, delirium y convulsiones.
ANSIOLÍTICOS SEROTONINÉRGICOS.
MEDICAMENTOS.
Buspirona |
farmacocinética:
Rápida y completa absorción por vía oral (que disminuye cuando se administra con alimentos) (Mattila et al., 1982). Biodisponibilidad limitada por el primer paso metabólico. t.máx = 40-90 minutos. UAP = 95%; vida media = 2.5 (2-11) horas. Inicio de acción de hasta 7 semanas. Tiene metabolismo hepático por hidroxilación; metabolitos activos: 5-hidroxibuspirona y 1-pirimidil-piperazina [1-PP] (que posee 1/4 de la actividad de la molécula original, pero más por efecto antagonista a2 noradrenérgico a nivel del locus coeruleus que serotoninérgico, lo que posiblemente contribuye al empeoramiento del trastorno de pánico cuando se administra esta medicación) (Tollefson et al., 1991). Eliminación renal: 29-63% y fecal: 18-38% (Eison & Temple, 1986).
farmacodinamia:
Agonista de receptores 5-HT1A somatodendríticos en las neuronas serotoninérgicas del rafe medio disminuyendo las tasas de disparo espontáneas de éstas (Meller et al., 1990), lo que conduce a la disminución de serotonina y sus metabolitos en el núcleo estriado, el hipocampo y el septum. La Buspirona puede también hiperpolarizar células piramidales hipocámpicas inhibiendo su disparo. El efecto acumulativo de reducción de los impulsos serotoninérgicos al hipocampo y áreas límbicas y corticales parece explicar los efectos comportamentales de las azapironas en algunos modelos animales de ansiedad (Taylor, 1989). La Buspirona tiene también una moderada afinidad por receptores D2, puede elevar los niveles de prolactina y probablemente de la hormona del crecimiento (Riblet et al., 1982 ; Meltzer et al., 1983).
Tiene efectos similares al de los antagonistas del GABA (diferentes a las BZD) (Skolnick & Paul, 1982) e incrementa la actividad de neuronas noradrenérgicas en el locus coeruleus (diferente a las BZD); por lo tanto, tiene poco efecto sobre la función psicomotora o cuando se asocia al alcohol, sin producir síndrome de abstinencia, ni dependencia física (Moskowitz & Smiley, 1982 ; Mattila et al., 1982 ; Cole et al., 1982); carece de efecto sedante (1/3 menor que las BZD), anticonvulsivante y relajante muscular, al activar la noradrenalina (Riblet et al., 1982 ; Eison & Temple, 1986).
indicaciones:
Tratamiento alternativo a las BZD del trastorno de ansiedad generalizada cuando se presenta marcada sensibilidad a los efectos sedativos (con efectos similares al Diazepam, Clorazepato, Alprazolam y Lorazepam) (Goldberg & Finnerty, 1979 ; 1982 ; Feighner et al., 1982 ; Schweizer et al., 1982 ; Rickels et al., 1982 ; 1988), pero es de elección en pacientes donde los síntomas ansiosos son de tipo cognitivo más que somático (siendo en esto muy superior al Diazepam y al Clorazepato) : hostilidad, rabia, preocupación, dificultad en la concentración, depresión y fatiga (Rickels et al., 1982 ; Goldberg & Finnerty, 1982) ; o en pacientes ansiosos con enfermedades pulmonares [Buspirona ha sido dada a pacientes con apnea del sueño con una disminución significativa en el número de apneas por hora y mejoría en todas las medidas de sueño (Mendelson et al., 1991) y no deprime la respuesta ventilatoria hipercápnica (al CO2) como si lo hace el Diazepam] (Rapoport et al., 1991).
A pesar de la adecuada sustentación de la eficacia de la Buspirona como ansiolítico en numerosos estudios controlados, para muchos clínicos sigue existiendo la duda sobre si el fármaco es igual de eficaz a las BZD. Esto posiblemente se debe al progresivo inicio de su actividad (debe esperarse la respuesta en un período de 3 a 4 semanas (Pecknold et al., 1989)), a que su eficacia es dependiente de la dosis (la cual debe ser titulada hasta llegar a niveles de 20 mg./día como mínimo) y a que el paciente tiene poca adherencia a un tratamiento en el que se requieren varias tomas al día. Sin embargo, el trastorno de ansiedad generalizada es de curso crónico y recurrente, lo cual exige una terapia prolongada en la mayoría de los casos con el consabido riesgo de abuso o adición a las BZD y de un síndrome de abstinencia ante la suspensión abrupta, lo cual no ha sido evidenciado con Buspirona. Un estudio muestra que un 65% pacientes que fueron asignados al grupo de Clorazepato seguían experimentando la necesidad de consumir el ansiolítico al cabo de 40 meses, versus ningún paciente asignado al grupo de Buspirona (Feighner, 1987 ; Rickels & Schweizer, 1990).
Puede usarse concomitantemente con BZD en casos resistentes, insomnio severo y al inicio, mientras se logra la respuesta a la Buspirona (Udelman & Udelman, 1990), pero teniendo en cuenta que la respuesta subjetiva a la Buspirona en estos pacientes será menos favorable que la de aquellos que nunca han recibido BZD.
La eficacia de la Buspirona puede incrementarse con el tiempo; un estudio abierto a largo plazo en pacientes con trastorno de ansiedad generalizada, mostró incremento de la mejoría clínica de un 50% a los 3 meses a un 70% a los 6 meses, aunque con una tasa de recaídas elevada (Feighner, 1987); sin embargo, la suspensión del medicamento puede no requerir adición de otras sustancias o su reiniciación hasta por varias semanas (Rickels et al., 1988).
Por el efecto noradrenérgico de su metabolito activo, la 1-pirimidinilpiperazina, puede tener eficacia en pacientes con alcoholismo y alto nivel de ansiedad (en la detoxificación y en la prevención del consumo), incluso en forma directamente proporcional a los niveles sanguíneos de dicho metabolito (Kranzler & Myers, 1989 ; Tollefson et al., 1991). Incluso, ha sido asociada a reducción de la ansiedad común en pacientes alcohólicos, que en algunos casos los lleva a la recaída posterior a la abstinencia (Kranzler et al., 1994). El consumo de cigarrillo se puede disminuir también, a pesar que el paciente no exhiba síntomas de ansiedad previos al inicio del tratamiento ; el paciente puede experimentar reducción de la ansiedad de abstinencia y de la fatiga (Gawin et al., 1989).
En un estudio placebo controlado doble ciego, se evidenció un efecto antidepresivo superior al placebo (65% vs. 28%) con dosis promedio de 57 mg. / día en 143 pacientes con depresión mayor y al menos moderadas cantidades de ansiedad (HAM-A > 15) (Rickels et al., 1991) ; sin embargo, debido a la ausencia de publicaciones que comparen Buspirona con un antidepresivo es poco prudente recomendarlo para tal fin. Otros autores encuentran apropiado el uso de la Buspirona en el manejo de la depresión refractaria como medicamento adicional al antidepresivo convencional, ya que se ha demostrado su efecto incrementador de la respuesta (Jacobsen, 1991 ; Joffe & Schuller, 1993). Una tercera opción, con pocos trabajos que la sustenten es el uso de la Buspirona en depresión ansiosa, donde parece ser efectiva no sólo en los síntomas ansiosos, sino también en los depresivos a dosis altas (Fabre, 1990 ; Rickels et al., 1991).
Otra indicación sugerida es el tratamiento de la discinesia tardía, extrapiramidalismo y acatisia inducidos por antipsicóticos, con altas dosis de Buspirona (entre 80 y 180 mg./día), posiblemente debido a la disminución de la supersensibilidad de los receptores D2 a través de su efecto agonista parcial (D'Mello et al., 1989 ; Moss et al., 1993). Su efecto es similar al de la Amantadina para controlar la catalepsia inducida por antipsicóticos (Goff et al., 1991).
La Buspirona puede tener efectos favorables en pacientes agitados con demencia (Tiller et al., 1988 ; Gelenberg, 1994), lesión cerebral (Yudofsky et al., 1990 ; Gualtieri, 1991), autismo (Realmuto et al., 1989) o retardo mental (Ratey et al., 1989 ; 1991).
El estudio comparativo de Liebowitz et al., reveló que la Buspirona había sido efectiva en el control de la fobia social en dos tercios de los 80 pacientes que ingresaron al estudio ; en el mismo, el Atenolol no fue superior al placebo. Las dosis van de 30 a 60 mg / día (Liebowitz et al., 1991 ; Schneier et al., 1993).
la Buspirona es poco eficaz para bloquear los síntomas de abstinencia por BZD, e incluso podría incrementar la ansiedad ; los efectos ansiolíticos de la Buspirona son menores en pacientes con historia de descontinuación reciente de BZD (Schweizer et al., 1982). En el TOC los resultados son divergentes ; Pato et al. encontraron que los pacientes tratados con Buspirona (60 mg./día) exhibían tasas de respuesta (reducción del 20%) similares a los que tomaban Clomipramina (Pato et al., 1991). Otros autores la descubrieron eficaz como medicamento adicional a la Fluoxetina (Markovitz et al., 1990 ; Jenike et al., 1991). Sin embargo, estos estudios no han sido replicados o han sido cuestionados por otros donde incluso se menciona empeoramiento del cuadro clínico (Jenike & Baer, 1988 ; Tanquary & Masand, 1990 ; Pigott et al., 1992 ; McDougle et al., 1993 ; Grady et al., 1993).
dosis:
15-30 mg/día (60 mg./día) divididos en 3 tomas. Iniciar con 5 mg. 3 veces al día y luego adicionar 5 mg. cada dos días hasta alcanzar dosis de 15 mg. 3 veces al día. Este proceso puede tomar unas 2 semanas y se lleva a cabo con el fin de disminuir los efectos adversos.
efectos adversos:
Seis efectos adversos son estadísticamente de mayor incidencia con Buspirona que con placebo : Mareo 30 minutos después de la ingestión (15%), náuseas (8%), cefalea (6%), nerviosismo (5%) e insomnio (2%). Otros efectos menos frecuentes son la disforia o sedación a dosis mayores de 30 mg/día que mejoran con el tiempo de administración, excitabilidad, fotopsias, parestesias, sudoración, boca seca, diarrea, taquicardia, dolor pectoral, confusión, depresión mental, efectos neurológicos y musculoesqueléticos, fiebre, visión borrosa, disminución de la concentración, bostezos, tinnitus, irritabilidad gástrica, pesadillas, fatiga... (Newton et al., 1982 ; Gelenberg, 1994 ; USP-DI, 1997). Al ser comparada con las BZD, todos los estudios coinciden en que no se presenta trastorno alguno en las funciones cognitivas en el rango terapéutico con Buspirona, ya sea en pacientes ansiosos o en voluntarios sanos (Moskowitz & Smiley, 1982). En un estudio comparativo se demostró que mientras el Lorazepam causaba un significativo y sostenido trastorno en las habilidades motoras, el equilibrio y la memoria, Buspirona sólo produjo un breve y modesto efecto en la memoria (Sellers et al., 1992).
interacciones medicamentosas:
No dar con IMAO's por el riesgo de hipertensión arterial (Ciraulo & Shader, 1990a). No dar concomitantemente con Digoxina, Ciclosporina o Disulfiram (riesgo de manía ?) (Iruela et al., 1991). Al darse con Diazepam, incrementa los niveles plasmáticos de Desmetildiazepam en un 20%. Su administración concomitante con antipsicóticos puede disminuir los efectos extrapiramidales de éstos, pero puede incrementar sus niveles plasmáticos a un 26% como en el caso del Haloperidol (Baughman, 1994). Puede reversar la disfunción sexual causada por los ISRS (Goff et al., 1991 ; Gelenberg, 1994), sin que se presenten interacciones peligrosas entre estos compuestos por la acción bimodal de la Buspirona; incluso puede potenciar el efecto antidepresivo de los ISRS (Joffe & Schuller, 1993).
precauciones:
No es carcinogénica, ni mutagénica, ni teratogénica, ni adictiva (Cole et al., 1982). Riesgo en embarazo: Categoría B. Puede excretarse por leche. Debe tenerse cuidado en pacientes con insuficiencia hepática o renal.
Tandospirona |
farmacodinamia:
Posee una alta afinidad y selectividad por receptores 5-HT1A. Se une tanto a los receptores somatodendríticos como a los postsinápticos con la misma afinidad. Actúa como un agonista completo de los autoreceptores 5-HT1A somatodendríticos y como agonista parcial de los postsinápticos (Godbout et al., 1991).
indicaciones:
Potente ansiolítico y débil antidepresivo (distimia). Eficacia similar a la Buspirona a pesar de poseer un mayor efecto serotoninérgico (Eison, 1990).
FUENTE: Tamayo JM. "Psicofarmacologia On-Line" http://psicofarmacologia.info/. Jorge M Tamayo, psiquiatra y farmacólogo clínico de la Universidad de Antioquia (Medellín, Colombia).